
What is Compliance?
Compliance is one of those buzz words thrown around in the Sterile Processing world that can mean a lot of things. You’ll often hear it used interchangeably with Accreditation, and leaders will be chattering about it leading up to the hospital’s accreditation survey. In this sense, compliance refers to healthcare organizations seeking to “comply” with the Conditions of Coverage for CMS, a state licensing department, or an approved accrediting organization. In this 3CE-approved education series, we will be looking at the standards and guidelines that Sterile Processing should be aware of, with particular attention given to manufacturers’ written instructions for use. Grab your SPD Leader, Infection Preventionist, and Accreditation Committee chair for this one, as we take a deep dive into the people, policies, and practices that make up a compliant Sterile Processing Department.

Guidelines That Drive Compliance
Compliance begins with awareness, since a department without standards and guidelines is as likely to be compliant as an archer with a blindfold on is successful at hitting a bullseye. There are three main categories of standards and guidelines that a Sterile Processing department needs to be aware of and adhere to:
- Regulatory – These standards are issued by regulatory agencies that wield the power and penalty of law. Examples include the Bloodborne Pathogen Standard issued by OSHA, requiring employers to provide adequate PPE to protect employees, the pre-market clearance for medical devices required by the FDA, and chemical use and disposal standards required by the EPA.
- Voluntary – These guidelines are developed and issued by professional organizations as best practices for the industry. These documents should direct the work practices of a Sterile Processing department, especially considering accrediting organizations will expect organizations to reference and abide by published best practices. The primary document Sterile Processing should be aware of is AAMI ST79, along with other standards from AAMI, AORN, AST, CDC, etc. For more info about AAMI standards, visit https://www.aami.org/standards
- Manufacturers’ IFUs – As an important subset of regulatory standards, manufacturers’ IFUs represent a significant portion of a department’s requirements for compliance. Developed by the manufacturer and cleared by the FDA, the IFU includes the validated instructions for reprocessing of that medical device. As such, Sterile Processing departments are required to reprocess the device in accordance with its validated instructions.

Training and Competency: Hardwiring Compliance
Expecting every Sterile Processing Tech to be an expert on all the applicable guidelines, standards, and IFUs would be an untenable ask. With so many standards to keep up with (and so many new Sterile Processing Technicians), how can a department possibly achieve compliance? Not to imply that it is easy (because it isn’t), here are some categorical actions that every Sterile Processing Department should take:
- Standard Procedures and Competencies – Departmental practice should be universally agreed upon by the Sterile Processing staff. There should be no question about what PPE to put on, how to brush an instrument in decontamination, how to load a sterilizer cart, and where sterile instruments may be stored. Departmental policies should be written out in accordance with industry guidelines, and staff member competencies should verify knowledge of and adherence to those policies and procedures.
- Initial Orientation – New technicians will always be a reality of Sterile Processing staffing, and so a strong training program that walks through department policies and procedures and verifies (and documents) competence at the end of precepting is required.
- Make Resources Available – Even if each technician does has not read the AAMI ST79 standards cover to cover, they need to at least have access to it. Team members should be able to demonstrate that they know where to go to get the answer to a question, particularly related to an instrument IFU. Here are some of the things you should keep available in your department for technicians:
- Industry Guidelines (AAMI, AORN, TJC Booster Pack, etc)
- Equipment Operator’s Manuals for all equipment
- Manufacturer IFUs for all instruments (physical or digital)
- As a bonus: certification study material for Sterile Processing Techs (HSPA or CBSPD)

Leading a Compliant Department
Central to a compliant Sterile Processing Department is an active and engaged leader. Without the influence of a leader shaping the culture of the department, the tide of convenience and apathy will undoubtedly sweep the department away into taking shortcuts, dropping consistent documentation, and ignoring difficult-to-apply best practices. As a leader, here are some things you can do to shape a culture of compliance:
- Patients First – Leaders set the tone for the day’s work, but they also set the mission for the journey. Shape the culture around patient safety, and empower technicians to be able to stand up for patient safety. This means that quality of instrument cleaning and inspection takes precedence over processing speed. It means that no shortcuts will be taken with instrument reprocessing steps to satisfy the pressing demands of the OR. Lead your department in doing the right thing, every time.
- Be Active – Leaders who are involved in the work have a better idea of “what is really going on.” Any leader can write a policy and give a training on it during a staff meeting. It takes an invested and engaged leader to be present during the workflow, see the obstacles and struggles the technicians face, and ensure that the work is being done according to policy in a seamless, efficient manner.
- Perform Audits – Audits get a bad rap for being a negative activity: something you do to try to catch people doing the wrong thing. Call them “work verifications” if you want to, but the point is to ensure that the agreed upon standard is being followed in the real world when the pressure is on. Random (or scheduled) audits are a great tool for leaders to ensure the process change is sustained.

Compliant Documentation
It isn’t enough to have compliant process in your department, you also need to be able to prove it. As the saying goes, “If it isn’t documented, it didn’t happen.” Here are come key examples of documentation in a compliant department:
- Sterilizer Records – Whether on paper or digital, a compliant department needs to keep record of sterilizer load contents, with the sterilizer identifier, operator, date and time, sterilizer parameters, and results of sterilization process indicators.
- Environmental Readings – Temperature and Humidity needs to be monitored and recorded in all areas of the Sterile Processing Department, recorded at least daily. For the acceptable parameters for your department, refer to ANSI/ASHRAE for the year your HVAC system was installed.
- Equipment Performance Testing – Departments are required to keep record of any equipment performance testing that is done. All instrument washers should be tested each day they are in use to confirm cleaning efficacy. Sterilizers (not including table top sterilizers) should receive a DART test daily when in use and a biological indicator test at least weekly (preferably daily) on all cycle types used. These records must be complete and accurate.
- Preventative Maintenance – Equipment preventative maintenance should be performed and documented. This includes ongoing maintenance called out in the equipment user manual such as sterilizer chamber cleaning, washer spray arm maintenance, and ultrasonic cleaner basin cleaning.
- Employee Competencies – This often overlooked documentation is required by most accreditation organizations and is vital to keeping an organized, compliant department. Verify your team’s competent adherence to department procedures on an annual basis and keep a record of it in their employee file.

Accreditation Surveys and Surveyors
Accreditation surveys are an almost universal experience for Sterile Processing Departments. While accreditation is technically voluntary (in most states), it is so common among healthcare facilities because of the financial incentive tied to accreditation. In order to participate in and receive federal payment from Medicaid or Medicare, healthcare organizations must be certified by CMS or by a contracted accrediting organization who applies the Medicare Conditions of Coverage requirements. Additionally, many private insurers are now requiring accreditation for payment.
Accreditation organizations conduct scheduled surveys on a cycle of every 3 years, with a possible interim survey if there were any deficiencies found that need to be rechecked. Random audits can be conducted at any time, announced or unannounced, by an accrediting organization. Surveyors will have training in Infection Prevention and some sterilization and disinfection and will be evaluating the Sterile Processing Department for adherence to policies and established best practices. Expect for surveyors to request to see documentation (such as employee files and sterilizer records), to speak with staff members about their job tasks, to observe work practices and environmental conditions, and to read policies and procedures. Deficiencies noted are weighted depending upon their severity and how widespread the issue is within the organization.

What is an IFU?
According to AAMI ST79, an Instructions for Use is written recommendations that have been provided by the manufacturer that “provide instructions for the operation and the safe and effective use of its device” (AAMI ST79, page 7). You’ll find these IFUs on a small piece of paper included with your medical device or instrument, and you can expect to find important information in it about how the instrument should be used, any limitations the device may have for reprocessing (such as a single-use device), and instructions for reprocessing the instruments.
Instructions for Use documents are important to Sterile Processing professionals, because those validated instructions are the only tested and proven steps and parameters that with achieve a sterile product. By following the steps that have been validated by the manufacturer, the user can have a high degree of confidence that the device is sterile and safe for use. If the Instructions for Use are not followed, the user assumes the risk associated with sterilization or device failure, since the FDA-approved validated reprocessing steps were not followed. Reprocessing instruments outside the parameters of the IFU also presents an accreditation risk, as surveyors expect departments to be compliant with manufacturer’s written instructions.

How are IFU's developed?
Medical devices, from idea to post-launch assessment, are regulated in the US by the FDA. The requirements for the medical device to be cleared or approved (known as the regulatory pathway) is dependent upon three device classifications, which indicate the degree of regulatory control needed to ensure its safety and effectiveness.
- Class I – Low-risk devices, like handheld surgical instruments, that must meet “general controls”, including registration, labeling, and quality system regulation. These are exempt from pre-market notification approval.
- Class II – Intermediate-risk devices, like most sterilizers, require a 510(k) pre-market notification clearance in addition to general controls and performance standards.
- Class III – High-risk devices, like heart valves and pacemakers, require a premarket approval (PMA) from the FDA that demonstrates safety and efficacy.
For all these medical devices, an IFU is required as part of the user communication. Good design practice incorporates an understanding of how the device will be used and by whom, and writes an instructional IFU that communicates information about storage, use, disposal, and reprocessing. Reprocessing instructions are to be based upon a validated process.

The IFU Knowledge Deficit
Despite all the labor and money that goes into writing and reviewing device IFUs, the sad reality is that the majority of Sterile Processing Techs will never get the time or chance to read it. They follow the standard procedures of their department, maybe not even aware that IFUs exist for all these instruments. If they have a question about how to wash a particular scope, they go ask their tenured coworker rather than turning to the IFU. The co-worker shares the “way we’ve always done it,” and they move on.
Situations like these are not uncommon, at they present a significant patient safety risk. If devices are not cleaned and sterilized appropriately, patients could be at risk of Surgical Site Infections (SSI) due to sterilization failure. As such, IFU non-compliance can result in an accreditation citation at the level of “Immediate Jeopardy”. The IFU for every surgical instrument and device should be kept on file in Sterile Processing where it is accessible. Whether paper copies are kept in an organized file cabinet, or the hospital subscribes to a digital library of IFUs, technicians should have the ability to access device IFUs when needed and the capability of reading and interpreting those IFUs.

Old News: Outdated IFU's in Sterile Processing
Device IFUs are updated over time by manufacturers for a variety of reasons. Technicians who are relying upon an old paper copy of the instrument IFU in a file cabinet may be using outdated information, calling into question the cleanliness and sterility of a device that has been processed using outdated IFUs. It is not enough just to have the IFU’s on hand, it is imperative that the SPD leader also maintain an up-to-date inventory of IFUs.
To accomplish this, healthcare organizations must first establish a policy that no instrument will be processed without a current IFU. This means that when vendors bring in instruments for processing and say, “just do it like you do everything else,” the department must insist on having a current IFU before processing them. Every time an instrument is purchased, the IFU should be compared to the one on file to verify no changes have been made. The facility could also utilize a digital library of IFUs that is automatically updated with the current version. If there is ever a question about which version of the IFU is current, contact the manufacturer directly.
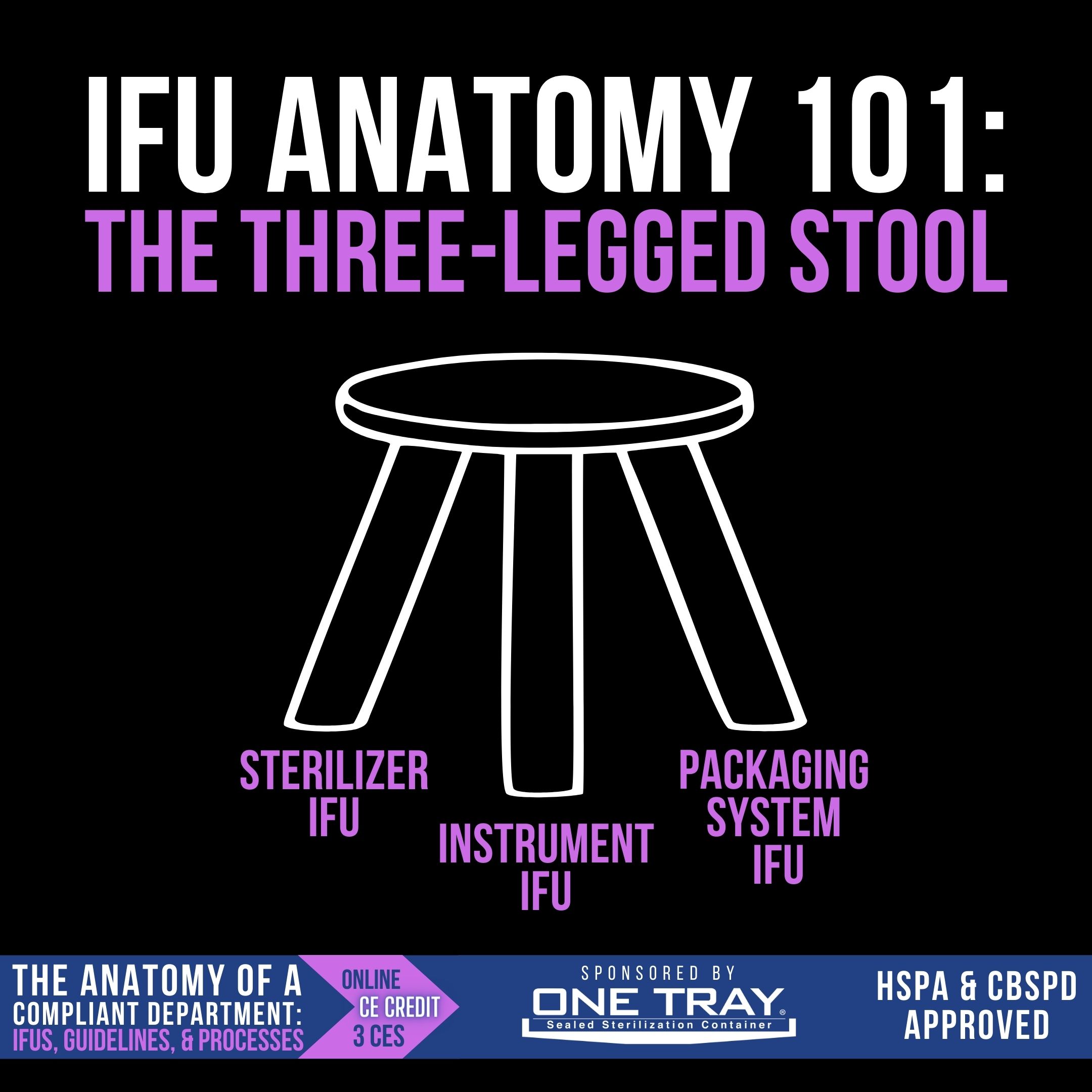
IFU Anatomy 101: The Three-Legged Stool
When reading instructions to determine how an instrument should be processed, there are several IFUs that need to be consulted: the sterilizer IFU, the instrument IFU, and the packaging system IFU. All three of these work together, like the 3 legs of a stool, to inform the user on how to sterilize and store those instruments. Let’s look at each leg individually:
- Sterilizer IFU – Rarely read by technicians in its entirety, the sterilizer IFU tells users how many trays can be sterilized in one load, the total weight allowed in a sterilizer load, the weight of individual trays allowed, how the packaging systems should be placed in the sterilizer, and the cycles that can be run.
- Instrument IFU – Included with the new instrument, the instrument IFU tells users the validated sterilization cycles, exposure time and exposure temperature, required to sterilize the device.
- Packaging System IFU – Whether wrap or rigid container, the packaging system IFU informs the users of the required dry times for the device and the permissible storage conditions and timeframe.
Each of these three IFUs inform the SPD technician’s decision-making for sterilization and sterility maintenance.

IFU Anatomy 201: How to Read an IFU
Manufacturer IFUs include important “how to” information for instrument use and reprocessing, as well as warnings and other device-specific information. There is no perfect guide for reading and interpreting IFUs, since every IFU is a little different, but here are some general guidelines for reading a medical device IFU:
- Warnings, Contraindications – The beginning of the IFU will usually include information about the device itself, unrelated to its use or processing. Information in this section that could be significant to Sterile Processing would be the instrument’s expected life cycle, sometimes measured in the allowed number of reprocessings.
- PreTreatment, Cleaning – Look in this section for specific instructions for point-of-use treatment, as well as steps to take in decontamination. IFUs will often specify soak times, volume of fluid to be flushed through lumens, number of brushings, etc. If automated cleaning is allowed, there are often still manual cleaning steps required.
- Sterilization – There is usually a table or chart in this area of the IFU with acceptable sterilization parameters for the different validated sterilization modalities. Select a sterilization cycle that meets these minimum requirements; over-sterilization is also not recommended. Read the text in this area for information regarding the use of various packaging systems (rigid containers, wrap, peel pack), number of reprocessings allowed, and Immediate Use Steam Sterilization instructions.
- Storage – Find instructions regarding sterility maintenance here, such as shelf life.

IFU's and Packaging Methods
During IFU development in the validation stage, the FDA only requires the instrument to be validated in one packaging method. This is significant because you are not necessarily required to use the same packaging method that the manufacturer validated the device in. AAMI ST79 makes room for users to choose another sterilization method, but they should conduct a sterility verification test (using the same sterilization exposure time and temperature) to confirm sterility can still be achieved using the new instrument configuration.
As referenced in the IFU Anatomy 101 post, dry time is a function of sterility maintenance, dictated by the packaging method manufacturer. As such, an instrument IFU may have a dry time listed in the sterilization section based on the packaging method used during validation testing. It is possible, if a facility were to use a different packaging method to sterilize the item, that the dry time would need to be extended or even shortened to match the new packaging method. The facility would, of course, still need to document a sterility verification test. This will be discussed shortly in the “Validation vs. Verification” post.

Developing Internal Policies, Including Defining "Immediacy"
A significant part of compliance is having policies that match your practice. Policies should be based upon industry best practice, be reviewed regularly, and be made available to staff. Each stage of the reprocessing cycle should be evaluated separately for conformance to best practices and have policies and standard procedures defined to standardize those processes. Here are two important policies you should make sure are in place in your facility:
- Sterility Assurance – How will your organization determine the shelf life of surgical instruments. According to industry guidelines like AAMI ST79, the sterility of terminally sterilized instruments can be considered event-related, meaning that they are considered sterile as long as they are kept in a controlled environment and protected from contaminants and mishandling. If the facility were to choose expiratory dating, the instruments would each have an expiration date.
- Instrument Reprocessing – A facility should state that they will follow instrument manufacturer’s IFU for reprocessing, and that no instrument will be sterilized without an IFU. This policy could also have a statement clarifying that sterilization dry time will be determined by the packaging method IFU, not the instrument IFU. This policy could also define “Immediacy”, as AAMI ST79 and other guidelines allow for each facility to establish their own policy on immediacy. A statement on immediacy could read something like this: “Immediacy is recognized when sterilization involves the use of a packaging system that cannot be stored for future use.”

Validation vs. Verification
The terms validation and verification are often confused and used interchangeably in the Sterile Processing world, but there is an important distinction between them. Validation is what is performed by a manufacturer to prove that a process will consistently yield a product that meets their specifications. A sterilization validation study proves that a specific set of sterilization parameters consistently yield a sterile product. Other types of validations can also be performed, like a cleaning validation. Users typically do not have the resources to perform a formal validation test in a healthcare organization that would meet the requirements of the FDA.
Verification is what is done by the user in their own environment. A user verification test establishes that the validated process can achieve its desired result in the real world. An example of this would be sterilizing a set of instruments (using your own rigid container and your own sterilizer), including several process monitoring devices like biological indicators and chemical indicators, and recording and documenting the results. The most common reason for performing a sterility verification test would be some reconfiguration of instruments, whether using a different arrangement of instruments in the tray or using a different packaging method.

No Two Are Alike: Non-Standard IFU Design
One of the primary reasons that there is such a knowledge deficit regarding IFUs in the Sterile Processing context is because of how varied they are. Unlike SDS sheets, which have a uniform format with identical sections, the manufacturer IFUs do not have to conform to a particular format or design. This means that every IFU a user reads will have unique features, unique language, and a unique flow. This non-standard design makes it very difficult for SPD professionals to quickly reference the specific information they are looking for.
Likewise, the design of the individual instruments in most loaned vendor sets are quite different, even though they may be used for the same purpose. Instruments that have a similar form and function from different manufacturers may have wildly different cleaning and sterilization instructions in the IFU. One may be designed to be disassembled, while the other remains assembled through the cleaning and sterilization process. Each instrument set needs to be researched separately.

The Overwhelming Quantities of IFU's
When you consider all the different manufacturers of instruments, different vendor instrument sets, and unique devices that are reprocessed by Sterile Processing, there are thousands of IFUs that SPD technicians are expected to understand and follow. One instrument set will have dozens of instruments in it, meaning that each of those IFUs must be referenced for reprocessing. For vendor instrument sets, each system typically has its own IFU, and it is not uncommon for medium-sized organizations to receive 50+ vendor trays in a day. With the sheer volume of IFUs needing to be kept and referenced daily, it is no wonder that Sterile Processing Departments struggle to be compliant in this area.
The majority of healthcare facilities are still tracking IFUs manually and do not have access to a digital IFU library. With this process, it is nearly impossible to be able to reference all the needed IFUs without grinding the workflow to a halt. For facilities that do not have any IFUs on hand, beginning to collect them and organize them feels like emptying the ocean with a thimble. However, IFU compliance is at the center for Sterile Processing’s mission and is a key component of compliance and keeping patients safe.

IFU's are Hard to Read
In addition to IFUs each having their own structure, layout, and design, their language is also varied. Many of them are complicated and filled with scientific jargon that can be difficult for the average SPD technician to interpret. Others are on the opposite end of the spectrum and are so simple and vague that they are unhelpful. They say things like “Use a hospital grade disinfectant…” or “Sterilize according to facility protocol.” Neither of these gives clear direction for the technician to follow and leaves the facility with more questions than answers.
Another problem with IFU variations is the wide variety of sterilization parameters used during validation studies. While the majority of instruments follow the industry standard of 270° F for 4 minutes with a dry time of 30 minutes, some manufacturers will pick exposure times as high as 20 minutes, with exposure temperatures from 250° to 273°. Cleaning instructions may be in metric measurements, referencing liters and milliliters instead of ounces and gallons.

Where to Find and IFU
One of the benefits of our time is the advent of social media groups and online forums where the Sterile Processing community can share ideas, best practices, educational information, and network with one another. One of the potential pitfalls of these forums, however, is taking the advice of a fellow SPD professional from one of these sites as authoritative truth. If you have a question about the reprocessing of a medical device, your only reliable source for instructions on reprocessing that item will be the manufacturer’s IFU.
There are several places you can find the IFU. If you have access to a digital library of IFUs online, you can search for it using the instrument title, manufacturer, and catalog number. If you don’t have access to such a library, many times the manufacturer website will have a portal for you to be able to look up and download a digital copy of the IFU. If neither of these options work for you, reach out to the manufacturer’s technical service line; they will usually be able to email you a copy of the sterilization instructions. If you have a question regarding the interpretation of the IFU, contact the manufacturer and request the clarification from them in writing.

"Off-Label" Reprocessing: When IFU's are not Followed
This program has addressed the importance of compliance in Sterile Processing, the challenges Sterile Processing technicians face in getting access to, maintaining, and interpreting IFUs, and what policies and procedures to put in place to ensure IFUs are being followed. But what about when IFUs are not followed? Here are three significant implications:
- Patient Safety Risk – This risk is the most significant, as IFU non-compliance can cause real harm to patients, potentially altering lives or even causing death. Failure to disassemble a complex vendor instrument during decontamination could easily redeposit bioburden into the next patient’s total knee surgical site and cause serious damage from the infection.
- Legal Risk – When a manufacturer validates instructions for cleaning and sterilization, they are assuming the risk for that device achieving the expected sterile outcome. When the facility processes the instruments according to a different process, they are assuming the risk related to the sterility of that instrument. When it comes to IFU’s the saying it true: “If you think compliance is expensive, try non-compliance!”
- Regulatory Risk – In addition to the risk to patients and the financial risk to the organization, failure to follow IFUs puts the department at risk for citation from an accrediting organization. Depending on the severity of the deficiency, the department could be shut down and surgeries cancelled until the deficiency is corrected.
